
- Синдром Крузона – что это?
- Общие сведения
- Виды заболевания
- Причины синдрома Крузона
- Патогенез
- Симптоматика
- Глазные расстройства
- Лицевые пороки развития
- Ротовые и верхнечелюстные мальформации
- Неврологические и нейропсихологические изменения
- Характеристика синдрома Крузона
- Осложнения и последствия
- Диагностика синдрома Крузона
- Лечение
- Медикаментозное лечение
- Операция
- Прогноз
- Профилактика
Синдром Крузона – что это?
Впервые эта патология была описана в 1912 г. Заболевание изучал французский педиатр О. Крузон, за что изнасилование получило одноименное название. Данная патология формируется во время внутриутробного развития. Чтобы понять, что это такое, важно понимать, что черепно-лицевые дизостозы – это большая группа дефектов, проявляющихся выраженным изменением формы лица и сопровождающихся гипертелоризмом. Синдром Крузона подпадает под эту особенность. Кроме того, для этого заболевания характерны и другие особенности, например, аномалии орбит.
Общие сведения
Синдром Крузона (краниофасциальный дизостоз 1 типа) — генетическое заболевание, характеризующееся нарушением процессов окостенения и развития скелетных элементов лицевого и мозгового черепа. Впервые это состояние было описано в 1912 г французским педиатром О. Крузоном, с тех пор синдром носит его имя. Механизм наследования синдрома Крузона аутосомно-доминантный, но заболевание часто вызывается спонтанными мутациями. Патология встречается достаточно редко: около 1,6 случая на 100 000 новорожденных, при этом почти 5% всех пороков развития, сопровождающихся краниальным дизостозом, обусловлены этим синдромом. Долгое время считалось, что это состояние имеет две разновидности: нормальное и сопровождающееся кожными нарушениями (гиперкератоз, акантоз),но с учетом современных данных специалисты в области генетики установили, что синдром Крузона с черным акантозом (КАН) представляет собой отчетливое наследственное заболевание. В то же время патогенез его развития аналогичен классической форме заболевания, чем и объясняется большое сходство симптомов. Заболевание с одинаковой вероятностью может затронуть как мальчиков, так и девочек.
Виды заболевания
Основным признаком развития патологии является деформация черепа, изменение его формы. Деформационные процессы могут быть разными, в зависимости от этого выделяют следующие формы заболевания:
- тригоноцефалия. У ребенка увеличен затылок и аномально узкий лоб;
- скафоцефалия. Отмечается удлинение черепа, узкий лоб. Череп ребенка низко посажен;
- брахицефалия Череп ребенка укорочен, голова слишком широкая;
- гидроцефальная форма. Череп младенца напоминает клевер.
Причины синдрома Крузона
Классический вариант синдрома Крузона обусловлен мутациями в гене FGFR2, расположенном на хромосоме 10, который кодирует аминокислотную последовательность рецептора 2 фактора роста фибробластов. Этот ген имеет значительный размер и большое количество экзонов, что снижает его стабильность : часто развиваются дефекты, которые приводят к многочисленным генетическим заболеваниям, поражающим в основном элементы скелета. Таким образом, кроме синдрома Крузона, мутации FGFR2 могут вызывать синдромы Аперта, Сетре-Чотцена, Бера-Стивенсона, Пфайффера и многие другие патологии. Генетические исследования показали, что краниофасциальный дизостоз 1 типа может вызывать более 35 мутаций вышеуказанного гена, которые в основном локализуются в области экзонов 7 и 9.
Почти все дефекты в гене FGFR2 представляют собой нонсенс-мутации, то есть вызывают изменение структуры кодируемого белка. Конформационное изменение рецептора 2 фактора роста фибробластов нарушает межклеточные взаимодействия в соединительных тканях черепа, прежде всего в костях и хрящах. Это приводит сначала к скоплению фибробластов в области межкостных швов, а затем к активизации процессов окостенения, что и является причиной основного проявления синдрома Крузона — черепного синостоза. Некоторые исследователи считают, что эти генетические дефекты влияют и на эмбриональное развитие структур первой жаберной дуги: к ним относятся челюсти и в некоторой степени элементы средней трети лица. Этим объясняется гипоплазия челюстей,особенно нижний, при синдроме Крузона.
Причины синдрома Крузона при черном акантозе несколько иные: он обусловлен мутациями в гене FGFR3, расположенном на 4-й хромосоме. Продукт его экспрессии также является рецептором фактора роста фибробластов, только 3-го типа (в отличие от 2-го типа, который является продуктом гена FGFR2). Только одна мутация этого гена оказалась причиной синдрома Крузона с характерными кожными проявлениями: Ala391Glu. Это также миссенс-мутация, которая изменяет структуру рецепторного белка. Патогенез заболевания практически не отличается от классического варианта. Изменения лица и черепа при синдроме Крузона с черным акантозом сходны с предыдущим типом, но к ним присоединяются гиперкератоз различных участков кожи и акантоз.
Патогенез
Патогенез синдрома прост: заболевание приводит к мутации гена фактора роста фибробластов FGFR2. Этот ген расположен на определенной хромосоме (10q26) и состоит из 20 участков с информацией о генах. Изменение, приводящее к возникновению синдрома Крузона, чаще всего локализуется в областях седьмого и девятого генов.
В целом ген FGFR2 может содержать 35 мутационных изменений, влияющих на развитие синдрома. Чаще всего такое нарушение возникает по отцовской линии.
У всех детей раннего возраста имеются швы — небольшие промежутки между элементами костей черепа и лица. По мере роста и развития ребенка расширяется головной мозг и благодаря этим швам происходит соответствующее расширение черепа. Шовные пространства срастаются только тогда, когда мозг полностью сформируется и перестанет расти.
У детей с синдромом Крузона швы заживают намного раньше, чем это необходимо. Таким образом, растущий мозг вынужден «вписываться» в доступное пространство. Внешне это заметно по нестандартной форме черепа, лица и зубных рядов.
Симптоматика
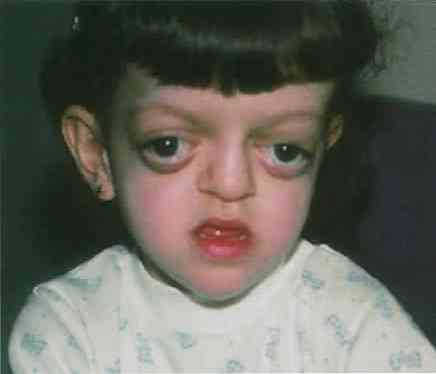
Клинические признаки синдрома Крузона весьма специфичны. Они заметны сразу после рождения. Тяжесть проявлений достигает максимума ближе к 3-4 годам.

- Основным признаком патологии является краниосиностоз, при котором кости черепа прочно сращены по венечному или стреловидному шву. При этом рост головы прекращается, и ребенок приобретает характерные черты: дистальный прикус, большое расстояние между глазами, клювовидный нос, низко посаженные уши, высунутый язык, укороченная и низкая губа, недостаточное смыкание челюстей, редкие зубы, высокое небо, выступающий вперед подбородок, расщелина языка. Характерным признаком синдрома является неполное смыкание зубов.
- Дополнительными признаками являются синдактилия пальцев, атрезия хоан, сколиоз, лордоз, мигрень, акантоз.
- По мере прогрессирования синдрома формируется короткая голова — брахицефалия. Возможные виды деформации черепа: клиновидная или ладьевидная головка, клеверолистная гидроцефалоидная деформация черепа. При пальпации определяются уплощенные края швов.

- Поражение зрительного анализатора проявляется расходящимся косоглазием, нистагмом, эктопией, колобомой. Экзофтальм обусловлен малой глубиной орбит. У больных с косым положением глаза постоянно вращаются, совершая спонтанные движения. Острота зрения значительно снижена. Частичная атрофия зрительного нерва выявляется у 80% больных.
- Нарушение слуха вплоть до полной глухоты возникает при поражении пирамиды височной кости, фиксации и деформации слуховых косточек. У больных изменяется форма внутреннего слухового прохода, снижается звукопроводимость костей, развивается атрезия наружного слухового прохода.
- Поражение нервной системы: умственная отсталость, задержка речи, судороги. Интеллектуальная недостаточность сохраняется на протяжении всей жизни, что затрудняет межличностное общение в обществе.
Негативные последствия болезни:
- гидроцефалия – водянка головного мозга,
- потеря зрения является результатом сдавления зрительного нерва и его атрофии,
- язвенный кератит развивается при неполном смыкании век и частичном подсыхании роговицы,
- психическое расстройство,
- трудно адаптироваться в обществе.
Глазные расстройства
Офтальмологическая область является одной из наиболее пораженных при синдроме Крузона, некоторые из наиболее распространенных патологий могут включать:
- экзофтальм: костная структура глазниц развивается с небольшой глубиной и поэтому глазные яблоки представляют собой переднее положение, т е как бы выпячиваются из этих полостей.
- Последствия кератита: аномальное положение глазных яблок вызывает большее обнажение их структур, из-за чего обычно возникает значительное воспаление глазных структур, расположенных в передних отделах..
- конъюнктивит: Как и в предыдущем случае, обнажение глазных структур может вызвать развитие инфекций, таких как конъюнктивит, вызывающий воспаление соединительной ткани..
- Гипертелоризм глаз: у некоторых людей может наблюдаться значительное увеличение расстояния между двумя глазами.
- Расходящееся косоглазие или экзотропия: в этом случае может наблюдаться отсутствие симметрии или параллелизма между обоими глазами, то есть когда один или оба глаза отклоняются в сторону латеральных областей.
- Атрофия зрительного нерва. Также может наблюдаться развитие прогрессирующей дегенерации нервных окончаний, отвечающих за передачу зрительной информации из областей глаз в головной мозг.
- нистагм: у некоторых людей постоянно наблюдаются непроизвольные движения глаз с аритмичным и быстрым проявлением.
- Катаракта: в этом случае хрусталик глаза становится непрозрачным и поэтому препятствует прохождению света к хрусталику для лечения. Пострадавшие люди будут иметь значительные нарушения зрительных способностей.
- Колобома радужной оболочки: может быть частичное или полное отсутствие радужной оболочки, то есть окрашенной области глаз.
- Нарушение зрения: у значительной части больных наблюдаются значительные нарушения зрительных способностей, во многих случаях это может протекать в виде слепоты различной степени тяжести.
Лицевые пороки развития
- Лобный отек: одной из наиболее характерных особенностей синдрома Крузона является наличие выпуклого или выступающего лба. Структура лобной кости имеет тенденцию к аномальному росту вперед.
- Пороки развития носа: в некоторых случаях можно наблюдать нос в виде «клюва попугая», то есть с опущенным или опущенным кончиком носа.
- Гипоплазия средней трети лица: в этом случае наблюдается частичное или замедленное развитие центральных областей лица.
Ротовые и верхнечелюстные мальформации
- Гипоплазия верхнечелюстных костей: у значительной части людей они представляют собой маленькую или недоразвитую верхнюю челюсть.
- Нижнечелюстной прогнатизм: Эта патология характеризуется выпуклостью или тенденцией к выходу из нижней челюсти, то есть находится в более выдвинутом положении, чем верхняя челюсть..
- Расщелина неба: в некоторых случаях может наблюдаться неполное закрытие крыши неба, в том числе губной структуры.
- Зубная окклюзия: смещение зубов или изменение положения прикуса является одним из наиболее частых проявлений в верхнечелюстной и щечной области.
Неврологические и нейропсихологические изменения
Пороки развития черепа могут препятствовать нормальному и экспоненциальному росту структур головного мозга и, следовательно, приводить к вариабельному наличию различных аномалий, таких как:
- Головная боль и рецидивирующая головная боль.
- Эпизоды судорог.
- Умственная отсталость.
- Прогрессирующая гидроцефалия.
- Повышение внутричерепного давления.
Характеристика синдрома Крузона
В частности, эта патология была впервые описана в 1912 году хирургом французского происхождения Октави Крузон (Бельтран, Росас-и-Хорхес, X).
Уже в первых клинических случаях, описанных в медицинской и экспериментальной литературе, можно было обнаружить четкую связь черепно-лицевых признаков с аномальным формированием черепных швов (Бельтран, Росас и Хорхес, X).
Самые последние заявления об этой патологии определяют ее как генетическое заболевание, возникающее в результате краниосиностоза или раннего закрытия костей, образующих череп (Genetics Home Reference, 2016).
Конфигурация черепа на младенческой или стадии развития имеет овальное строение, расширяющееся в задней области. Так, костные отделы (затылочный, височный, теменной и лобный) обычно формируются на пятом месяце гестации и представлены вместе соединительной или фиброзной тканью, черепными швами (Villareal Reyna, 2016).
Следовательно, черепные швы позволяют расширять голову и мозг благодаря своей гибкости. Кроме того, его закрытие начинает постепенно развиваться между 9 и 24 месяцами (Villareal Reyna, 2016).
При изменении этого процесса, например краниостенозе, происходит раннее закрытие этих фиброзных структур (Villareal Reyna, 2016).
Таким образом, это мероприятие препятствует нормальному формированию строения черепа, лица и головного мозга. Как следствие, у пораженного человека развивается множество пороков развития, которые влияют на глаза, положение челюсти, форму носа, зубы или формирование губ и неба (Genetics Home Reference, 2016).
Хотя у большинства людей с синдромом Крузона нормальные или ожидаемые потребности для их возрастной группы, нормальное развитие мозга может замедляться и, как следствие, могут возникать различные трудности в обучении, которые вместе с аномальными зубами и зубами верхней челюсти значительно замедляют овладение языком. Домашний справочник по генетике, 2016 г).
В дополнение к наиболее часто используемому термину «синдром Крузона» эта патология может также иметь отсылки к другим типам названий: краниостеноз типа Крузона, черепно-лицевой дизостоз или черепно-лицевой дизостоз Крузона (Национальная организация по редким заболеваниям, 2007)..
Осложнения и последствия
Синдром Крузона не может пройти бесследно: как правило, у ребенка возникают различные последствия и осложнения:
- гидроцефалия;
- ухудшение зрения, вплоть до потери (из-за длительного сдавления зрительного нерва в нем происходят необратимые изменения);
- истончение и язвенное поражение роговицы (из-за чрезмерного выпячивания глазных яблок веки становятся невозможными для полного смыкания, в результате чего роговица частично подсыхает и покрывается язвами);
- умственная отсталость;
- трудности с адаптацией в социуме (психическая неполноценность и неприятные внешние проявления синдрома существенно затрудняют взаимодействие больного с социумом).
Еще одним осложнением синдрома может быть аномалия Арнольда-Киари – это перемещение миндалин мозжечка через большое затылочное отверстие в шейные позвонки.
Диагностика синдрома Крузона
В первую очередь врач осматривает больного ребенка. Он может уточнить, происходило ли это в семье, ведь признаки синдрома Крузона достаточно характерны и их трудно спутать.
Инструментальная диагностика, которая проводится в обязательном порядке сразу при подозрении на синдром, поможет врачу уточнить диагноз.
Рентгенограмма покажет стадию закрытия ламбдовидного, венечного и стреловидного швов. Кроме того, этот метод помогает выявить уменьшение придаточных пазух носа, признаки базилярного кифоза, увеличение гипофизарной ямки, неправильную форму глазниц.
Топографически наблюдается деформация внутреннего слухового прохода. Кроме того, на топограмме висков прослеживается наружная ротация каменистой части пирамиды, имеющая место на фоне дисплазии основания черепа. Визуально это проявляется гиперостозом, косой ориентацией слуховых проходов, аномальным ходом лицевого нерва.
КТ или МРТ подтверждают следующие признаки:
- атрезия;
- сужение наружного слухового прохода;
- деформация камер сосцевидного отростка и стремени;
- отсутствие барабанной полости;
- анкилоз молоточка;
- нарушение развития периостальной части лабиринта.
Дополнительно врач может направить пациента на консультацию к другим специалистам, которые на свое усмотрение назначат анализы и другие исследования. Например, при подозрении на синдром Крузона уместны консультации генетика, психиатра, невропатолога, офтальмолога, нейрохирурга.
Лечение
Синдром Крузона — неизлечимое заболевание, требующее функциональной и косметической коррекции. Этого можно добиться только хирургическим путем.
Хирургическое вмешательство направлено на восстановление формы черепа и устранение синостозов. Такие операции проводят с рождения до прекращения роста черепа. Они позволяют снизить уровень внутричерепного давления, исправить деформации опорно-двигательного аппарата, предотвратить дальнейшее прогрессирование умственной отсталости, а также нервно-психических расстройств. Для этого раскроют слишком большие швы черепа и подкорректируют форму лица.

Основные виды операций:
- Краниопластика и раскрытие синостотических швов: удаление и расширение костных фрагментов черепа для предотвращения повреждения головного мозга и коррекции формы черепа.
- Создание искусственного блефарофимоза для уменьшения выраженности экзофтальма и коррекции положения глазного яблока.
- Расширение хоан для восстановления дыхания.
- Установка вентрикулоперитонеальной шунтирующей системы при гидроцефалии.
- Использование аппарата Илизарова для перемещения верхней челюсти вперед и улучшения функции дыхания.
- Коррекция выступающей нижней челюсти и нормализация ее внешнего вида.
- Радикальные комплексные операции направлены на устранение дефектов лица: исправление недоразвития верхней челюсти и восстановление зубного ряда.
В настоящее время в хирургическую практику внедряются дистракционные методики коррекции деформаций костей лица. С помощью специальных приспособлений для смещения любой части черепа и коррекции костных дефектов процесс лечения проходит быстрее и эффективнее.

В случаях, когда операция ребенку противопоказана, нарушение функции внешнего дыхания компенсируют постоянной оксигенотерапией, дыханием под высоким давлением, применением воздуховодов и трахеотомией.
Медикаментозная терапия также поможет облегчить состояние больного. Больным назначают:
- Ноотропы – «Пантогам», «Пирацетам», «Винпоцетин».
- Сосудистые препараты – «Кавинтон», «Циннаризин», «Актовегин».
- Диуретики – «Лазикс», «Верошпирон», «Диакарб».
Больные с синдромом Крузона находятся на учете у офтальмологов и отоларингологов, которые при выявлении отклонений назначают соответствующую терапию. Дети с психическими расстройствами нуждаются в специальном лечении и логопедии.
Медикаментозное лечение
В зависимости от симптомов, наблюдаемых у ребенка, могут назначаться различные лекарственные препараты, в том числе:
- Средства, улучшающие кровообращение и укрепляющие сосуды. Они способствуют нормализации мозгового кровообращения и состояния сосудов головного мозга.
- Ноотропные препараты способствуют стимуляции питания клеток головного мозга и поддержанию их нормальной функции.
- Антиконвульсанты останавливают эпилептические припадки.
- Нестероидные противовоспалительные препараты уменьшают воспаление и снимают боль.
- Осмотические диуретики, кортикостероиды нормализуют внутричерепное давление.
- Ретиноиды уменьшают тяжесть кожных пятен.
Операция
Операция делается для того, чтобы снять швы и дать мозгу возможность расти. После операции детям придется несколько месяцев носить специальный шлем, чтобы изменить форму черепа. Операция также может быть выполнена для:
- Ремоделирование черепа для исправления краниосиностоза. Операцию проводят нейрохирург и черепно-лицевой хирург. Изменения вносятся поэтапно и расширяются по мере роста мозга.
- Фронто-орбитальное продвижение. Это делается для увеличения пространства внутри черепа и размера обеих глазниц (части черепа, удерживающей глазное яблоко).
- Прогрессирование средней части лица. Операция проводится в возрасте пяти лет или раньше, если дыхание затруднено. Щеки и нижние орбиты глаз выступают. Можно использовать методы, стимулирующие рост костей.
- Бисептум лица. Он расширяет верхнюю челюсть, сужает глазницы и сужает верхнюю часть лица.
- Остеотомия Движение костей верхней и нижней челюсти для исправления дальнейших нарушений прикуса. Обычно это делается в подростковом возрасте.
- Ринопластика. Пластика носа.
- Гениопластика. Пластика подбородка или щек.
Дети с нарушениями слуха могут использовать слуховые аппараты для усиления звука. Это состояние может также потребовать речевой и языковой терапии.
Прогноз
Прогноз синдрома Крузона неоднозначен. Это зависит от выраженности деформации черепа и состояния мозговой ткани. Первичную реконструктивную операцию проводят на первом году жизни. По мере роста ребенка она дополняется новыми вмешательствами на костях и мягких тканях лица.

Заболевание часто сопровождается задержкой психомоторного развития и судорожными припадками. Поэтому многие специалисты оценивают прогноз как неблагоприятный. Даже при полноценной поддерживающей терапии у больных прогрессирует экзотропия, развивается глухота и гипоплазия лица. Пациенты, вовремя обращающиеся за медицинской помощью и находящиеся под постоянным наблюдением специалистов, часто доживают до преклонного возраста. Некоторые люди с синдромом Крузона все еще сохраняют социальную адаптацию, несмотря на серьезные косметические недостатки.
Причинами инвалидности являются нарушения зрения и слуха, а также умственная отсталость. Со временем костные дефекты становятся более выраженными. Поскольку профилактические меры по предотвращению развития этого заболевания еще не разработаны, остается только надеяться на прогресс медицинских технологий, которые однажды смогут избавить людей от генетических нарушений.
Профилактика
Профилактика рождения детей с синдромом Крузона невозможна, так как это заболевание в подавляющем большинстве случаев носит наследственный характер.
Поскольку иногда встречаются спорадические случаи синдрома, что может быть связано с преклонным возрастом отца ребенка на момент зачатия, рекомендуется тщательно взвешивать степень риска при планировании столь «поздней» беременности.
Если в семье уже были случаи рождения детей с синдромом Крузона, то родителям имеет смысл пройти полное обследование у генетика на наличие мутированного гена FGFR2.
Всем беременным, независимо от качества их наследственности, рекомендуется своевременно (не позднее 12 недель) встать на учет в ЖК, а также регулярно посещать гинеколога.